Confounding effects and extra tricks
Confounding effects and extra tricks
Getting started
We’ll continue using the same data from previous days.
Load packages
require(gdata)
require(metafor)
require(dplyr)
require(compute.es)
require(ggplot2)
require(cowplot)
require(pez)
require(phytools)
require(ape)
Download data (Curtis et al. 1999)
curtis<-read.xls("http://www.nceas.ucsb.edu/meta/Curtis/Curtis_CO2_database.xls",as.is=TRUE,verbose=FALSE,sheet=1)
curtis_ES<-escalc(measure='ROM', m2i=X_AMB , sd2i=SD_AMB, n2i=N_AMB, m1i=X_ELEV, sd1i=SD_ELEV, n1i=N_ELEV, vtype='LS',var.names=c("LRR","LRR_var"),data=curtis)
## Warning in log(m1i/m2i): NaNs produced
#summary(as.factor(curtis_ES$PARAM))
curtis_WT<-filter(curtis_ES, PARAM=="TOTWT") # let's use whole plant weight because it has the largest number of observations
curtis_WT$GEN_SPP<-paste(curtis_WT$GENUS,curtis_WT$SPECIES,sep="_")
Conversion among effect sizes
‘Compute.es’ is a powerful package that converts effect sizes.
The main function for calculating effect sizes is mes and des ,and res convert among effect sizes.
Here, we will first calculate basic effect sizes using mes and then convert the effect size r to Fisher’s z
# calculate effect sizes
curtis_ES<-mes(m.2=curtis_WT$X_AMB, m.1=curtis_WT$X_ELEV, sd.2=curtis_WT$SD_AMB, sd.1=curtis_WT$SD_ELEV, n.2=curtis_WT$N_AMB, n.1=curtis_WT$N_ELEV ,verbose=FALSE)
# convert correlation coefficient to fisher's z
new_ES<-res(r=curtis_ES$r,var.r=curtis_ES$var.r, n=curtis_ES$N.total,verbose=FALSE)
curtis_ESS<-dplyr::select(new_ES, r, var.r, N.total, fisher.z, var.z)
head(curtis_ESS)
## r var.r N.total fisher.z var.z
## 1 0.71 0.02 8 0.89 0.20
## 2 0.31 0.08 10 0.32 0.14
## 3 0.59 0.03 10 0.68 0.14
## 4 -0.21 0.09 10 -0.21 0.14
## 5 0.19 0.11 8 0.19 0.20
## 6 0.81 0.01 8 1.13 0.20
r_z<-ggplot(curtis_ESS, aes(x=r, y=fisher.z))+geom_point() +
geom_abline(intercept = 0, slope = 1,colour="red")+theme_bw()
r_z

hist_r<-ggplot(curtis_ESS, aes(r)) +
geom_density(colour="red")+xlab("r")+theme_bw()
hist_z<-ggplot(curtis_ESS, aes(fisher.z)) +
geom_density(color="blue")+xlab("Fisher's Z")+theme_bw()
ab<-plot_grid(hist_r, hist_z, ncol=2)
ab

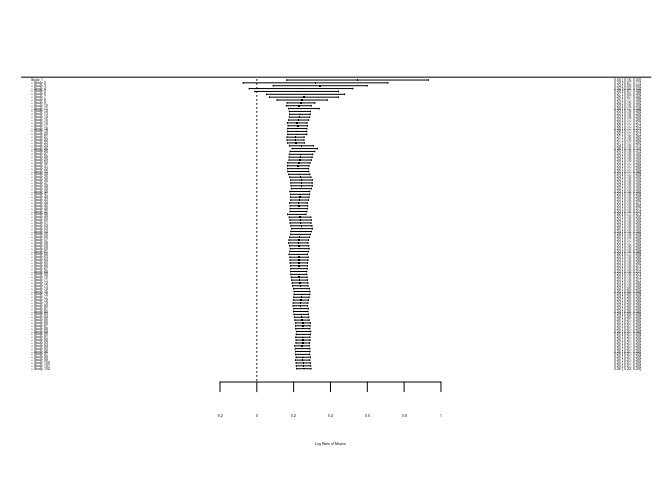
Cumulative meta-analysis
This method tests whether effect sizes have shifted over time. It fits the model by iteratively adding observations in the order that we designate.
re_wt<-rma(LRR, LRR_var, data=curtis_WT)
cum_re<-cumul(re_wt, order(curtis_WT$OBSNO))
forest.cumul.rma(cum_re)
Controlling for shared evolutionary history (phylogeny)
For species-level analyses, shared evolutionary history should be controlled for above and beyond random effect terms already included.
Clean data set
The basic steps are:
- Resolve taxonomic names to ensure that as many as possible can be placed on a phylogeny.
- Build phylogeny. If you’re interested in how to build a phylogeny, here’s the code
- Make sure that your phylogeny has the same number of tip labels as there are species in the data set.
setwd('/homes/dc78cahe/Dropbox (iDiv)/Teaching/MetaAnalysis_Course/pages/Day4_files/')
clean<-read.csv("TPL_sppnames.csv")
clean<-dplyr::select(clean,GEN_SPP2=Taxon, phy=new_species)
clean$GEN_SPP2<-as.character(clean$GEN_SPP2)
curtis_WT$GENUS<-tolower(as.character(curtis_WT$GENUS))
curtis_WT$GENUS<-paste(toupper(substr(curtis_WT$GENUS, 1, 1)), substr(curtis_WT$GENUS, 2, nchar(curtis_WT$GENUS)), sep="")
curtis_WT$SPECIES<-tolower(as.character(curtis_WT$SPECIES))
curtis_WT$GEN_SPP2<-as.character(paste(curtis_WT$GENUS, curtis_WT$SPECIES,sep=" "))
curtis_WT$GEN_SPP2<-ifelse(curtis_WT$GEN_SPP2=="Populusx euramericana","Populus × euramericana",curtis_WT$GEN_SPP2)
curtis_WT<-dplyr::left_join(curtis_WT,clean, by="GEN_SPP2")
# read in tree
tree<-read.tree("Curtis_phylogeny.tre")
str(tree)
## List of 5
## $ edge : int [1:68, 1:2] 36 37 37 38 39 39 40 40 38 41 ...
## $ Nnode : int 34
## $ tip.label : chr [1:35] "Pseudotsuga_menziesii" "Picea_glauca" "Picea_mariana" "Picea_abies" ...
## $ edge.length: num [1:68] 144.6 207.6 23.1 100.8 83.7 ...
## $ node.label : chr [1:34] "Spermatophyta" "" "" "" ...
## - attr(*, "class")= chr "phylo"
## - attr(*, "order")= chr "cladewise"
# we need to drop one species from our data frame ('Trichospermum mexicanum' because it wasn't placed on the phylogeny)
curtis_WTT<-filter(curtis_WT, phy!="Trichospermum_mexicanum")
length(unique(curtis_WTT$phy))
## [1] 35
#same number of species on phylogeny as in data set?
length(unique(curtis_WTT$phy))==length(unique(tree$tip.label))
## [1] TRUE
Fit multi-level meta-analytical model that accounts for shared phylogenetic history
- Make a phylogenetic correlation matrix
- Fit model such that there is a random term for species, which is then matched to a correlation matrix (‘R’)
- Compare it to a model that doesn’t account for phylogeny
tree_m<-vcv.phylo(tree, cor=TRUE) # creates phylogenetic correlation matrix
re_phy<-rma.mv(LRR, LRR_var, mods=~1,random=list(~1|PAP_NO, ~1|XTRT, ~1|phy), R=list(phy=tree_m), data=curtis_WTT)
summary(re_phy)
##
## Multivariate Meta-Analysis Model (k = 101; method: REML)
##
## logLik Deviance AIC BIC AICc
## -22.7040 45.4080 53.4080 63.8287 53.8291
##
## Variance Components:
##
## estim sqrt nlvls fixed factor R
## sigma^2.1 0.0124 0.1115 29 no PAP_NO no
## sigma^2.2 0.0080 0.0895 8 no XTRT no
## sigma^2.3 0.0088 0.0936 35 no phy yes
##
## Test for Heterogeneity:
## Q(df = 100) = 768.9932, p-val < .0001
##
## Model Results:
##
## estimate se zval pval ci.lb ci.ub
## 0.2745 0.0693 3.9597 <.0001 0.1386 0.4103 ***
##
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
#
re_nophy<-rma.mv(LRR, LRR_var, mods=~1,random=list(~1|PAP_NO, ~1|XTRT, ~1|phy), data=curtis_WTT)
summary(re_nophy)
##
## Multivariate Meta-Analysis Model (k = 101; method: REML)
##
## logLik Deviance AIC BIC AICc
## -20.4116 40.8232 48.8232 59.2439 49.2443
##
## Variance Components:
##
## estim sqrt nlvls fixed factor
## sigma^2.1 0.0087 0.0933 29 no PAP_NO
## sigma^2.2 0.0094 0.0969 8 no XTRT
## sigma^2.3 0.0042 0.0645 35 no phy
##
## Test for Heterogeneity:
## Q(df = 100) = 768.9932, p-val < .0001
##
## Model Results:
##
## estimate se zval pval ci.lb ci.ub
## 0.2742 0.0455 6.0271 <.0001 0.1850 0.3634 ***
##
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Accountring for phylogeny, in this particular case, did not alter the mean effect size. However, the confidence intervals around the mean effect size are wider when accounting for phylogeny.
Exercise: do your own meta-analysis (using either Gibson et al. or another data set of your own choosing)
Tasks
- Download data from Gibson et al.
- Calculate effect sizes
- Fit multi-levelr andom effects model
- Make visualisations of your results (effect sizes, forest plot, etc.)